Run Gromacs Faster on Rescale with Parallelization
Gromacs was developed by ScalaLife and funded by the European Research Council. Gromacs is an open-source, versatile package designed for analyzing molecular dynamics in biochemical molecules. Due to its popularity, we decided to use Gromacs to demonstrate the steps required to parallelize a single job across multiple dynos (i.e., computing cores). You can find the original basic Gromacs tutorial here
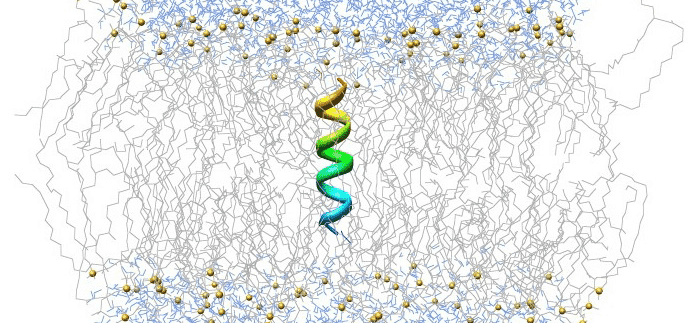
Our example simulates a phospholipid membrane, consisting of 1024 dipalmitoylphosphatidylcholine (DPPC) lipids in a bilayer configuration with 23 water molecules per lipid, for a total of 121,846 atoms. When you encounter large-scale problems, consider parallelizing your job across multiple dynos to reduce runtime. If you can successfully parallelize your job across two dynos, you could potentially reduce your job’s runtime by 50%.
When we parallelized our example across four dynos, the runtime was cut in half.

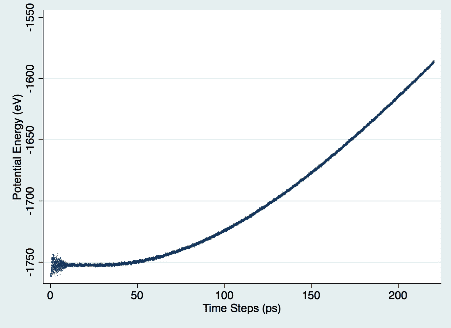
As you can see, we didn’t experience a 4x improvement in runtime when parallelizing across four cores (when compared to running on one core). There are delays associated with (a) job decomposition and (b) passing of messages between the various dynos involved in the job. In addition, if decomposition is not done well, the resulting load imbalance leads to additional delays.
Luckily, Gromacs provides users a time-accounting tool to keep track of all these factors. To use it, simply open the “job-name”.log file with a text editor:
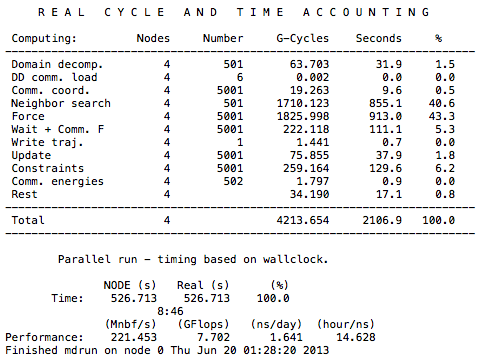
The time unit here is in CPU seconds. You can calculate the real time spent on each part of the job by dividing this value by the number of dynos involved, which, in this case, is four dynos. Domain decomposition, waiting, communication, and reading/writing are some of the factors that stop us from achieving ideal scaling. However, advanced users can use the data provided by Gromacs to learn from inefficient runs and design more efficient parallelizations.
If you want to learn more about Gromacs on Rescale, contact us at support@rescale.com